
基石药业-B(02616)8项重磅临床研究进展亮相学术大会
本文来自微信公众号“医药魔方”,文中观点不代表智通财经观点。
基石药业-B(02616),一家专注于开发及商业化创新肿瘤免疫疗法及精准治疗药物的领先生物制药公司,在本月召开的两场国内外肿瘤学会(CSCO、ESMO)大会上披露了多个重磅产品的最新研究进展,包括抗PD-L1单抗舒格利单抗(CS1001)以及抗PD-1单抗CS1003、FGFR4抑制剂Fisogatinib和GIST领域靶向创新药阿泊替尼等,其研究领域主要集中在胃腺癌或胃食管交界处腺癌、胃肠间质瘤、肝癌、淋巴瘤等重要瘤种和急需突破的治疗领域。这些研究数据凸显了基石药业强大的研发实力与丰富的产品管线,这些积极的研究结果都显示了在患者人群中具有良好的安全性和耐受性。
舒格利单抗(CS1001)
舒格利单抗是由基石药业开发的在研抗PD-L1单克隆抗体。作为一种全人源全长抗PD-L1单克隆抗体,舒格利单抗是一种最接近人体的天然G型免疫球蛋白4(IgG4)单抗药物。与同类药物相比,舒格利单抗在患者体内产生免疫原性及相关毒性的风险更低,这使得舒格利单抗在安全性方面具有独特的优势。近期,4项重磅研究进展数据以口头报告、壁报、摘要等形式亮相ESMO/CSCO大会。
CS1001-201研究
学术会议:CSCO
形式:口头报告
CS1001-201研究是一项评价舒格利单抗单药治疗复发或难治性结外自然杀伤细胞/T细胞淋巴瘤(R/R ENKTL)的单臂、多中心、II期、关键性研究。其主要研究终点为经独立影像评估委员会(IRRC)评估的客观缓解率(ORR),次要研究终点为经研究者评估的ORR、经IRRC和研究者评估的完全缓解率(CR)、中位缓解持续时间(mDoR)、无进展生存期(PFS)及总生存期(OS)等,并评估药物的安全性及药代动力学和免疫原性。
截至2020年7月1日,共计43例患者入组并接受治疗。入组患者的特征与ENKTL发病人群相似,74.4%患者的ECOG体力状态评分为1分;72.1%患者入组时疾病已处于IV期,约一半(51.2%)的患者已接受过二线及以上治疗。
舒格利单抗单药治疗R/R ENKTL患者,获得突出的CR率,持久的mDoR以及OS的获益,并具有良好的安全性和耐受性。
38例疗效可评估患者的ORR 为44.7%,其中CR率高达31.6%。mDoR为16.8个月,获得CR的12例患者中有11例仍在持续缓解中。
接受给药的43例患者的中位总生存期(mOS)达到了19.7个月,1年OS 率为55.5%
最常见舒格利单抗相关不良事件为发热;6例(14.0%)患者发生≥3级与研究药物相关的不良事件;10例(23.3%)患者发生免疫相关不良事件,多数为1级;3例(7.0%)患者发生了舒格利单抗相关严重不良反应(SAE)CS1001-101研究
学术会议:CSCO
形式:壁报
CS1001-101研究是一项针对晚期实体瘤或淋巴瘤患者开展的旨在评估CS1001的安全性、耐受性、药代动力学特征和抗肿瘤疗效的Ⅰ期研究。在本次CSCO年会上报道的是CS1001-101研究中舒格利单抗联合XELOX(卡培他滨和奥沙利铂)作为一线方案治疗无法进行手术的局部晚期或转移性胃腺癌或胃食管交界处腺癌(GC/GEJ)b期队列的疗效验证(POC)数据。研究主要终点为联合疗法的初步抗肿瘤疗效。
舒格利单抗联合XELOX 作为一线治疗方案在晚期GC/GEJ患者中具有显著的抗肿瘤活性,并且具有良好的安全性和耐受性。初步生物标志物分析显示PD-L1的表达水平与舒格利单抗联合XELOX的治疗效果具有潜在相关性。
截至2020年2月19日,在纳入有效性分析集的29例患者中,经研究者评估的ORR为62.1%,其中18例患者达到部分缓解,6例患者达到疾病稳定;mDoR为11.3个月;中位无进展生存期(mPFS)为8.3个月;mOS为17.0个月。
有效性分析集中的26位患者具有可评估的PD-L1表达联合阳性评分(CPS)结果。分析显示,CPS ≥ 5亚组(19例患者)的ORR为58%,mDoR尚未达到,mPFS为13.3个月;在CPS < 5亚组(7例患者)中,ORR为71%,mDoR为5.0个月,mPFS为6.2个月CS1001单药联合化疗一线研究
学术会议:ESMO
形式:壁报
舒格利单抗(CS1001)在ESMO会议上也带来了胃腺癌或胃食管交界处腺癌、食管鳞状细胞癌(ESCC)两个队列的临床研究数据。其中,胃腺癌或胃食管交界处腺癌数据与CSCO会议一致,而在食管鳞状细胞癌(ESCC)队列中,CS1001再次表现了有效的疾病控制率和客观反应率。
截至2020年2月19日,37例患者可进行有效性分析,有25(67.6%)例达到了PR,包括20例已确认PR和5例未确认PR;SD的患者有8例(21.6%);2例(5.4%)PD;2例(5.4%)患者在未达基线后的肿瘤评估时终止治疗(即不适用)。客观反应率ORR为68%, mPFS为9.0个月, mDoR和mOS仍未达到。
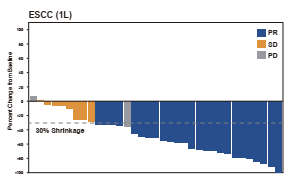
与CS1001相关的所有等级和≥3级不良事件(AEs)分别为87.2%和41.0%。最常见的(n≥2例)≥3级与CS1001相关的AEs包括贫血(n=6)、白细胞计数减少(n=3)、中性粒细胞计数减少(n=3)、血淀粉酶增加(n=3)、血小板计数降低(n=2)、低钠血症(n=2)和乏力(n=2)。
CS1001 群体药代动力学研究
学术会议:CSCO
形式:摘要
CS1001群体药代动力学研究是一项基于模型的群体研究,用于描述CS1001药代动力学特征并探索可能对其产生影响的协变量。
目前的群体模型共包括了168位患者的1146个观察数据,数据来源于两项实体瘤I期临床试验(CS1001-101,102)和一项淋巴瘤II期临床试验(CS1001-201)。该研究考察了人口学(年龄、性别、种族等),肝肾功能(谷丙转氨酶、谷草转氨酶、肌酐清除率等)及药物分子相关(抗药抗体)的一些变量对CS1001药代动力学特征的影响程度。
试验结果显示,CS1001药代动力学特征符合一级消除的二室动力学模型。基于模型,一名典型59.1kg,白蛋白为42.4g/L的男性患者,预测清除率和中央室表观分布容积约为0.19L/天和3.21L。协变量筛选研究显示,白蛋白和体重为清除率主要协变量,体重和性别为中央室表观分布容积的主要协变量。其余协变量包括种族、年龄、肝肾功能、抗药抗体未见对CS1001药代动力学有显著影响。模型评估显示,该模型可以准确预测CS1001的集中趋势和变异度,亦可被用于作为剂量选择及调整的相关证据。
阿泊替尼(avapritinib)
阿泊替尼是一种激酶抑制剂(c-Kit/PDGFRα),FDA于2020年01月批准其用于治疗携带PDGFRA基因18号外显子突变(包括PDGFRA D842V突变)的不可切除性或转移性GIST成人患者。此前,尚无针对该疾病的药物上市,接受伊马替尼和其它许可药物治疗的PDGFRA D842V突变型GIST患者预后较差,总缓解率为0%。此次CSCO大会披露的数据是阿泊替尼在中国人群中的I/II期初步桥接研究结果,研究显示阿泊替尼在中国患者中耐受性良好,且对携带 PDGFRA 外显子18 D842V 突变的中国患者具有很强的抗肿瘤活性。基石药业已于今年4月向CDE递交了这款药物的新药上市申请 (NDA),并获得优先审评资格。
阿泊替尼中国人群桥接研究
学术会议:CSCO
形式:口头报告
此项桥接研究是一项开放标签、多中心的I/II期临床研究,旨在评估阿泊替尼治疗不可切除或转移性晚期GIST患者的安全性、药代动力学特征和抗肿瘤疗效,并依据I期剂量爬坡研究的初步结果确定II期临床研究的推荐剂量(RP2D)。截至数据截止日期2020年3月31日,共计50例中国患者纳入阿泊替尼的安全性评估,8例携带D842V突变的患者以及23例4L+患者的疗效可评估,由研究者依据实体瘤疗效评价标准1.1 版 (RECIST)进行评估。
截至数据截止日期,6例使用阿泊替尼200mg 每日一次治疗,44例使用300mg 每日一次治疗。I期研究中,患者在200mg和300 mg剂量下对该药均显示出了良好的耐受性,研究中未观察到剂量限制性毒性。在中国GIST患者中的II期研究推荐剂量确定为300 mg,每日一次口服,这与针对晚期GIST全球研究NAVIGATORI期研究中的数据保持一致。
阿泊替尼在携带PDGFRAD842V 突变的患者中初步显示出了显著的抗肿瘤活性。在300mg每日一次的剂量下,8例携带PDGFRAD842V突变的患者中,所有患者靶病灶均有缩小,有5例患者达到了研究者评估的部分缓解,3例患者达到了研究者评估疾病稳定,总体缓解率(ORR)为62.5%。阿泊替尼在至少接受过3线既往治疗的(4L+)GIST患者中也显示出一定的疗效,研究者评估的ORR为26.1%。阿泊替尼总体安全性和耐受性良好,与全球其他研究结果一致。研究中报告的治疗相关不良事件(TRAE)大部分为 1 级或 2 级。最常见的治疗相关 TRAE 为贫血和血胆红素升高。最常报告的≥3级(均为3级)TRAE为贫血。
Fisogatinib(BLU-554)
Fisogatinib是一个高效高度选择性FGFR4抑制剂。FGF19通过与FGFR4和KLB结合后传导信号,异常的FGF19可能驱动肝细胞癌(HCC)的发生并造成预后不良,HCC中异常表达FGF19配体。Fisogatinib是基石药业引进的创新药物,本次CSCO大会披露的结果为fisogatinib单药治疗HCC的临床有效性数据,其疗效进一步证实了FGF19-FGFR4信号通路在肝细胞癌中的致癌驱动作用,也进一步证实了FGF19是fisogatinib治疗HCC的预测性生物标志物。此外,fisogatinib正在进行与CS1001(PD-L1抑制剂)联合在晚期HCC中的安全性及疗效研究。
Fisogatinib(BLU-554)
BLU-554-1101研究
会议:CSCO
形式:壁报
BLU-554-1101研究是一项评估BLU-554 在肝细胞癌患者中安全性、耐受性、药代动力学、药效学和初步疗效的I期试验。主要分为三个部分,第一部分主要确定药物的最大耐受剂量(MTD)和/或II期推荐剂量(RP2D);第二部分为在RP2D下的剂量扩展研究;第三部分为在既往未接受过酪氨酸激酶抑制剂(TKI)治疗的FGF19免疫组化阳性(IHC阳性)的HCC患者中进行的在RP2D(600 mg QD)剂量下的扩展研究。该研究的第一部分和第二部分结果表明fisogatinib在FGF19过表达的HCC中是可耐受并且有效的。本次CSCO大会披露了第三部分中11例中国患者的初步疗效和安全性数据。
疗效:Fisogatinib在FGF19IHC阳性的11例既往未接受过酪氨酸激酶抑制剂(TKI)治疗的晚期肝细胞癌中国患者中显示出了令人鼓舞的疗效。客观有效率(ORR)为36.4%(4/11,其中3例患者为确认的部分缓解(PR)),3例确认的肿瘤缓解深度超过60%且缓解持续。
安全性:Fisogatinib在中国肝癌患者中安全耐受可控可管理,没有4-5级治疗相关不良事件(TRAE)发生,27.3%(3/11)的患者发生了3级TRAE;没有不良事件(AE)导致的治疗终止;常见的TRAE为丙氨酸氨基转移酶(ALT)升高(90.9%)、腹泻(81.8%)、天门冬氨酸氨基转移酶(AST)升高(72.7%)和血胆红素增加(63.6%)。
CS1003(PD-1 单抗)
CS1003是基石药业通过领先的杂交瘤技术平台开发的一种针对PD-1的全长、人源化免疫球蛋白G4 (IgG4) 单克隆抗体,该药在临床前体内试验中已表现出了良好的耐受性和有效性。ESMO大会上,基石药业公布了CS1003的两项研究数据,分别为CS1003在晚期肿瘤患者的药代动力学特征、安全性、及初步抗肿瘤活性研究(CS1003-101 1b)和联合仑伐替尼(lenvatinib,LEN)用于一线治疗(1L)不可切除肝细胞癌(uHCC)中国患者的Ib期临床研究(CS1003-102 1b)。在这两项临床研究中,抗PD-1单抗CS1003不论单药还是联合标准治疗,均显示了良好的安全性与有效性。
CS1003-101研究
会议:ESMO
形式:壁报
CS1003-101研究Ib期部分探索CS1003不同给药方案(200mg,Q3W;400mg, Q6W)下的有效性、安全性、药代动力学研究,主要研究终点是研究者基于RECIST V1.1评估的客观缓解率,次要研究终点是为评估无进展生存期(PFS)、疾病控制率(DCR)、疗效持续时间(DOR)和总生存期(OS)、并评估药物的安全性及耐受性、药代动力学和免疫原性。
截至2020年7月15日,Cohort A(200 mg Q3W)和Cohort B (400 mg Q6W)分别入组29例和31例患者,并全部纳入有效性和安全性分析。两组客观缓解率(ORR)分别为24.1% 和 32.3%,其中,确认的客观缓解率(confirmed ORR)为20.7% 和 25.8%,中位响应时间两组均尚未达到。
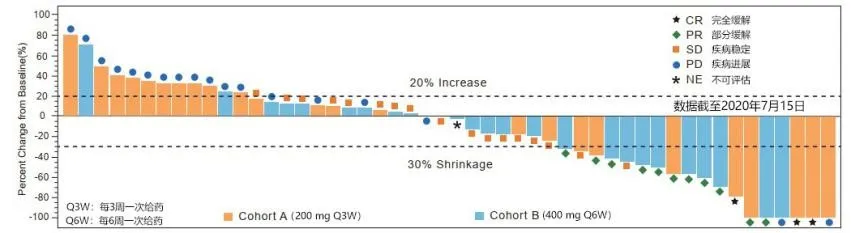
安全性方面,Cohort A(29例)和Cohort B(31例)患者至少发生一起治疗出现的不良事件在两组分别占患者总体的96.6%和96.8%,大部分治疗相关的不良事件都是1-2级,两组的3-4级的治疗相关的不良事件发生率分别为0和9.7%,未发生治疗相关的死亡,免疫相关的不良事件发生率两组基本相当(34.5% vs. 32.3%)。
Cohor t A和CohortB分别有15例和4例患者数据纳入有效稳态PK暴露量分析,稳态下的CS1003的平均血浆药物浓度(平均血药浓度,Cavg,ss)在两组间基本相当。因此,选择400 mg Q6W给药方案将为患者提供一种更灵活更便捷的给药方案选择。
CS1003-102研究
会议:ESMO
形式:壁报
CS1003-102研究是一项在中国开展的多中心、开放性的剂量递增和适应症扩展的I期研究,该研究第五组旨在评估CS1003 联合仑伐替尼(lenvatinib, LEN)一线治疗在中国不可切除肝细胞癌(uHCC)患者中安全性和有效性。主要研究终点为研究者基于RECIST V1.1评估的客观缓解率(ORR),次要研究终点为评估无进展生存期(PFS)、疾病控制率(DCR)、疗效持续时间(DOR)和总生存期(OS)、并评估药物的安全性及耐受性、药代动力学和免疫原性。结果显示,CS1003 联合仑伐替尼(lenvatinib, LEN)一线治疗在中国不可切除肝细胞癌(uHCC)具有较高的ORR且安全性良好
截至2020年6月22日,共招募20例患者并全部纳入有效性分析,8名患者在最终评价时确认为客观缓解,客观缓解率ORR达到40%(8/20),中位随访时间为6.2月,中位PFS为8.4月,截至数据截止日期,中位OS尚未达到,中位响应时间尚未达到。
20例患者最常见的治疗相关不良事件(TEAE)为血胆红素升高、尿蛋白阳性和蛋白尿。3级TEAE的发生率为25%(5/20),包括高血压、结合性胆红素升高、腹泻、糖尿病和低磷酸盐血症;未见报告4级及以上的TEAE。
(编辑:李国坚)


 扫码下载智通APP
扫码下载智通APP